Blog AI4Chem
Welcome to the blog!
Organisation
Clicking on the top-left hamburger icon  you can toggle the table of contents.
you can toggle the table of contents.
License
The prose is all under CC BY 4.0, external content is linked and if I find the license it is explicitly stated (in image captions, for example).
Tools Used
mdbook.- A few text objects (e.g. quotes, nav headings) use Computer Modern font;
- The rest of the content uses Ubuntu > System UI > sans-serif, in that order of preference.
Atom Vectors - Introduction
Atom vectors is a broad category. Narrower categories are:
- Descriptor: used for expert-designed vectors,
- Embedding: used for machine-learnt vectors.
These vector representations are of interest because they can be useful inputs for training machine-learning models.1
Here we are interested in methods that don't require human expertise, hence descriptors won't be discussed.
Embeddings
Embeddings are machine-learnt vectors . Normally, they are also:
- Dense rather than sparse i.e they have few zeros,
- Real-valued rather than discrete,
- Non-human-readable (though some parts may be interpretable),
- Belong to a structured vector space: semantically similar vectors are close together and allow for meaningful vector-arithmetic.
Dense vectors are useful because training will be faster (than a similar sparse version).
Some Background on Embeddings
Embeddings for atoms were inspired by NLP models from the 2010s.
One such example was learning continuous vector representations of words (2013). They proposed an automated mechanism generate word-vectors by absorbing information from that word's environment (neighbouring words).
Materials science has exploited the same ideas, for example:
properties of an atom can be inferred from the environments it lives in
(Atom2Vec, 2018),atoms are to compounds as words are to sentences
(SkipAtom, 2022),
The surprise was that similar words (or atoms) produce similar vectors. The vectors also support semantically meaningful arithmetic operations, and became useful for downstream tasks. A classic example was:
vector("Queen") = vector("King") - vector("Man") + vector("Woman")
Vectors in Chemistry
As dicussed earlier, atom vectors can be expert-designed or they can be learnt by an algorithm. Learning vectors yields more general-purpose vectors, and has won in popularity.
Both Atom2Vec (2018) and SkipAtom (2022) are unsupervised algorithms that obtain their atom vectors from databases of compounds. Atom vectors can be combined into compound vectors, and used for downstream tasks like property-prediction.
Classifications and Featurisers
The method used to generate our vectors is called a featuriser (we can use a featuriser or create our own). There are many common approaches:
- Simple: like hot-encoded, random;
- Human-designed: Composition-Based Feature Vector (CBFV) which are expert-curated vectors as in Jarvis, Magpie;
- Machine-learnt: embeddings, like SkipAtom.
Atom-vectors can be combined to describe compounds. Examples of combination methods are concatenation into a long vector and pooling of vectors (e.g. summing them up).
Comparing representations
A performance-comparison of vector representations is carried out in "Is domain knowledge necessary for machine learning materials properties?" (2020).
Their conclusion is: human-designed Composition Based Feature Vectors (CBFV like Jarvis and Olyinyk) outperform other methods if there isn't much data. This was prior to SkipAtom, but does include Atom2Vec.
Otherwise, performance in downstream tasks is similar to hot-encoded or random vectors.
(...) Although new, data-driven approaches are of interest, those studied here have yet to surpass CBFVs in terms of material property prediction with small data.
However, "Domain Independent XAI for Material Science" (2025) challenges that conclusion:
Our method challenges this perception: we obtain excellent classifiers that are interpretable and based on a small amount of training data without using any domain knowledge: (...)
They assert that one-hot encoded vectors can still achieve good results using small datasets, as long as the network is designed in the way they specify.
Thoughts
Human-designed vectors are easier to interpret. Machine learnt vectors require more effort. Can we design machine-learnt interpretable vectors that are intrinsically interpretable?
Attention-masks and disentangled representations are closer to this.
-
Machine Learning (ML) includes deep-learning and traditional methods like regression, support vector machines, random forests and so on. ↩
Atom Vectors - Atom2Vec
A popular representation of atoms as vectors appeared in (2018): Atom2Vec.
They take compounds from a database, and build a matrix like the one below:1
| 1 | 1 | 1 | 0 | 0 | 1 | 0 | |
| 0 | 1 | 1 | 1 | 0 | 0 | 1 | |
| ... | 1 | 0 | 1 | 0 | 0 | 1 | 1 |
Let's describe using the compound as an example:
- When is the target, it generates , placed in the first column.
(2)is the stoichiometry of the element in the compound. - When is the target it generates , placed in the fourth column.
(3)is the stoichiometry of the element in the compound.
Since a particular atom binds to a very small fraction of all groups, each row is very sparse (high fraction of zeros). The same is valid for columns.
SVD Method
A normalised matrix is obtained by normalising each row vector independently. Using the euclidean norm (2-norm) allows for an intuitive similarity metric:
In their best-performing model, they compute , collect the -rows with the largest singular values, and compute where is the slice of rows of D with the largest singular values, and the corresponding columns.
Note
The strategy has certain beauty to it: the new f-vectors retain the inner product similarity but are denser. Though now, the columns have no explicit meaning.
Findings
- Similar atoms have similar vectors,
- Increasing the distance threshold in stages, vectors can be clustered hierarchically, from the leaf-nodes (atoms) downwards (groups).
- At some level, groups match the periodic table groups. (I don't know how the grouping is made unambiguous).
- At a very large distance, all atoms merge into a single group. The result is called dendogram.
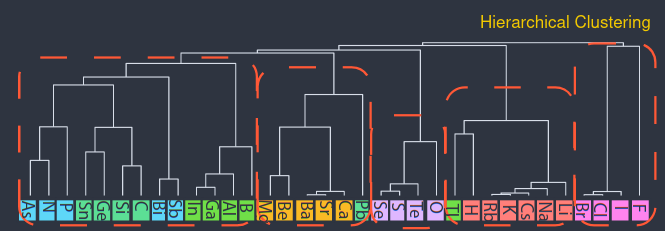
Image (modified) from Original Paper under CC-BY-SA 4.0. The atoms are rotated to make the image fit (rotated).
- Looking at the variation of some dimensions in the vectors, we can assign meaning to some of them.
Benches
Then, they compared to "empirical features" —a vector (group, period,...), padded to match their — with the task of predicting the DFT-found formation-energies of elpasolite crystals ().
Each solid was represented as a concatenation of atom vectors, and feed it to a hidden layer. (They also do other tasks.)
The paper ends with an interesting insight:
Structural information has to be taken into account to accurately model how atoms are bound together to form either environment or compound, where the recent development on recursive and graph-based neural networks might help.
-
It would be a binary matrix but the database contains some compounds multiple times and those are left duplicated (for some strange reason). ↩
Atoms Vectors - SkipAtom
Atom2Vec was already described; now it's time for SkipAtom, another algorithm to learn atom embeddings.
Method
First, compounds are downloaded. Then, the Voronoi Decomposition is used to derive graphs from unit-cells, and from the graphs generate training-pairs. As they show in the paper:
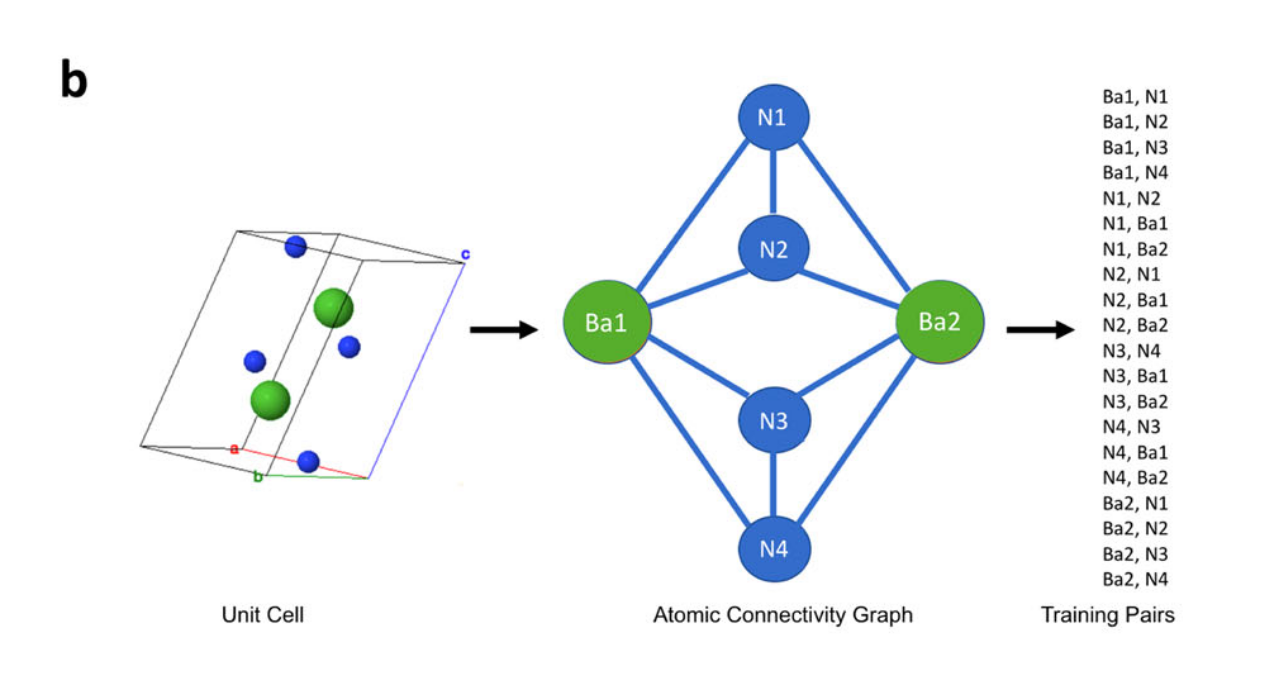
Image from Original Paper under CC-BY-SA 4.0
Finally, each pair X-Y is used to train a shallow network to predict the target (Y) from the reference (X).
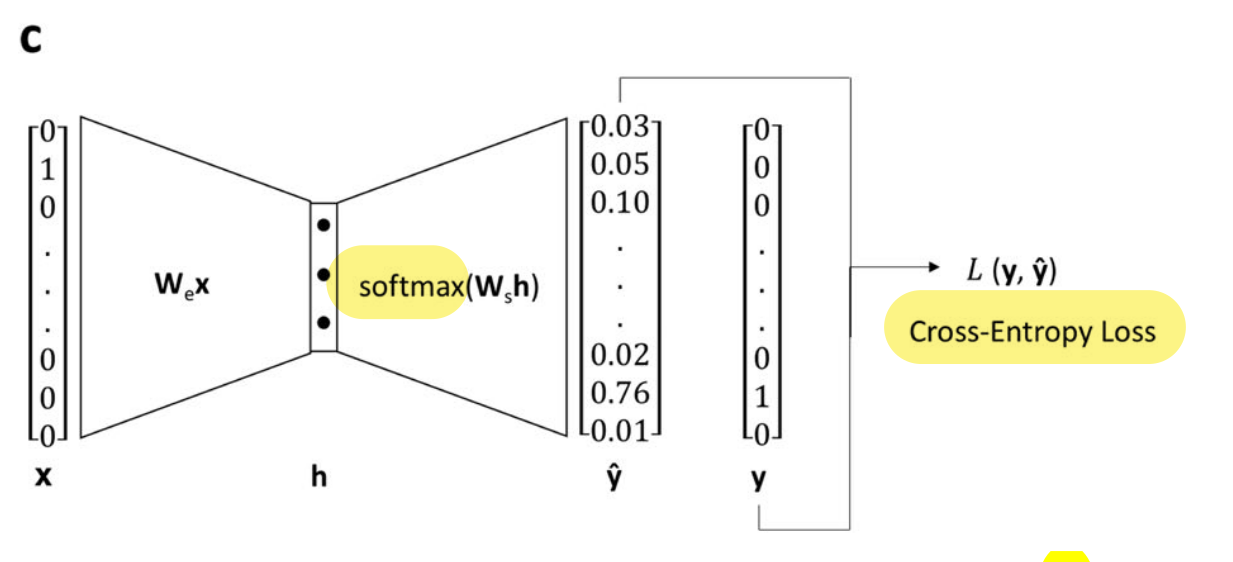
Image from Original Paper (slightly modified) under CC-BY-SA 4.0
- The resulting representation is dense and structured/semantic. This can be shown using dimensionality reduction techniques (PCA, t-SNE,..).
- The architecture is described as:
(...) single hidden layer with linear activation, whose size depended on the desired dimensionality of the learned embeddings, and an output layer with 86 neurons (one for each of the utilized atom types) with softmax activation. (...) minimizing the cross-entropy loss between the predicted context atom probabilities and the one-hot vector representing the context atom, given the one-vector representing the target atom as input.
Representations of Compounds (Pooling)
The analogy to NLP is that words are like atoms, and sentences are like compounds. Hence, distributed representations of atoms can be combined (pooled) into a vector representing a compound.
Vector-pooling options are:
- sum: where is the stoichiometry (can be fractional),
- mean: , i.e. divided by total number of atoms (can be fractional too).
- max: , reduces material matrix to vector. Selects max value of each column, each row being an atom in the compound.
The resulting compound representation is then used for training a feed-forward NN on different tasks. Also benchmarked using MatBench.
The pooling can also be done with hot-encoded vectors for atoms. This is done in ElemNet (mean pooling), and in Bag-of-atoms (sum pooling). The advantage: no training required, the disadvantage: the result is a sparse vector, and can be less accurate.
Results
They run two groups of tests:
- Embeddings' quality through elpasolite task. Where the atom vectors are concatenated into a compound vector (no pooling). The vectors train a network for property prediction. SkipAtom performs best here.
- Embeddings quality and pooling methods, through 9 prediction tasks. The results are:
- Pooling: sum and mean-pooling outperform max-pooling,
- Kind: Mat2Vec does best, and second SkipAtom,
- Bag-of-Atoms (sum pool of hot enc) does best in one task.
They conclude that these methods are most useful when no structural info is available.
However, there isn't a simple answer to which representation is best, it depends in the task. This is discussed in more detailed in Results.
Reference-based Coordinate Assignment
RCA is a dimensionality reduction method. Importantly, this implies we already have some representation available, which we may want to reduce.
The main idea could be conceptually represented as:
On the right hand side, the distance of each dimensional vector to is calculated. Each of these vectors is a centroid calculated from a cluster of embeddings.
The result, on the left hand side, is a reduced -dimensional sample vector .
An important point is raised in the paper:
To avoid degeneracies arising from equidistant configurations, the reference points must span the structure of the dataset.
Which means the reference vectors (centroids) should be able to reach any point in the original dataset, and not lose expressivity.
The method has useful properties:
- Compared to a large vector, it is cheaper to use to train a model (and can still be accurate),
- Compared to other dimensionality reduction methods, it is easier to extend,
- With certain care, the dimensions may be interpretable.
Comparison
SkipVec and Atom2Vec were previously discussed.
The paper Is domain knowledge necessary for machine learning materials properties? compared descriptors (same as vector representations) generated in different ways for downstream tasks.
They find hand-crafted descriptors useful for small and large datasets, but these are cumbersome to create —expert knowledge is required. One-hot and random-vectors perform similar to hand-crafted descriptors in large datasets.
With that, it seems wise to use hand-crafted descriptors for small datasets, and learnt, one-hot or random for larger ones.
SkipAtom evaluates on different approaches and tasks, and finds their method outperforms one-hot and random-vector, but does not test hand-crafted ones.
SkipAtom's comparison of representations is discussed below.
Simple classification
We have a simple classification of the available methods:
- Human-engineered vectors;
- Low effort vectors: one-hot encoded (ElemNet), random, Atom2Vec;
- Machine-learnt vectors (SkipAtom).
Atom2Vec may be in category 3 above; however, it is not an optimisation that creates them, it is a matrix factorisation.
Quality of Atom Representations
ElemNet (One-hot), Random, Atom2Vec, Mat2Vec and SkipAtom compared.
The atom-vectors were concatenated into compound representations, and these used to predict elpasolites (compounds) formation-energy.
SkipAtom outperformed other methods.
Pooling approaches
Vector pooling strategies were compared through 9 prediction tasks; 5 regressions, 3 classifications and OQMD Formation Energy prediction (also a regression).
- OQDM: bag-of-atoms, which is sum-pooling hot-enc atom vectors, is best.
The rest is summarised well in the paper:
(...) the models described in this report outperform the existing benchmarks on tasks where only composition is available (namely, the Experimental Band Gap, Bulk Metallic Glass Formation, and Experimental Metallicity tasks). Also, on the Theoretical Metallicity task and the Refractive Index task, the pooled SkipAtom, Mat2Vec and one-hot vector representations perform comparably [to the SOTA], despite making use of composition information only.
And an interesting observation:
The ElemNet architecture demonstrated (...) Perhaps surprisingly, the combination of a deep feed-forward neural network with compound representations consisting of composition information alone results in competitive performance when comparing to approaches that make use of structural information.
Use cases and limitations
Training does not rely on labelled data (unsupervised learning).
The model just needs the formula at inference time, and does fine with non-stoichiometric solids. So having the material's composition —but no structural information— we can still calculate some properties.
Similar compounds have similar vectors, which is useful. But without structural information, all isomers have the same vector, which is a limitation.
It is computationally cheap, and can help screen large number of compounds as a first selection step.
Explainable AI - Concepts
There are many definitions of what explanations are in the context of explainable AI, such as:
Explanations are answers to questions about the model's predictions or operation.
How does it operate? Why does it makes certain prediction? What is the role of certain neuron or layer?
In contrast to explainable models, black-box models share no insight about their operation or predictions.
Multiple Dimensions
The questions and the answers depend on the audience, the context and other dimensions.
One paper on XAI for radiology found that doctors preferred text outputs + saliency maps to aid their decisions (in a sense, emulating doctor-to-doctor communication). The output was then evaluated by a clinician.
Similarly, in synthetic chemistry, new candidates are vetted by expert chemists. Again, text can be a useful piece of information for the expert to work on, consider, evaluate. However, with no "human in the loop", this approach isn't enough.
The list of explanation dimensions may be infinite. A few are:
- Intend, Purpose, Stakes: such as transparency, trust, ethical reasons. Healthcare, finance, energy, military, general agentic-systems and automated decision-making.
- Yet in other scenarios, explainability may not matter —but only the model's output, which a human expert could evaluate.
- User Type / Audiences: Each audience has goals, risks, and preferences. Scientists, ML practitioners, developers, non-experts.
- Stages: pre-modelling, modelling, post-modelling.
- Kind: Visual, Textual, Formal. More below.
Yet another dimension related to the explanation kind and its complexity is recalled in The perils and pitfalls of explainable AI: Strategies for explaining algorithmic decision-making:
Belle and Papantonis (2021) provide four suggestions for creating explainability, including explanation by simplification, describing the contribution of each feature to the decisions, explaining an instance instead of in general, and using graphical visualization methods for explanations. At the same time, they also discuss the complexity of realizing such suggestions. Simplifications might not be correct, features can be interrelated, local explanations can fail to provide the complete picture, and graphical visualization requires assumptions about data that might not necessarily be true.
Disentangling Dimensions
We can consider characteristics of explanations independent of the audience and context:
- Simplicity: how easy to understand the explanation is. (The opposite term, complexity, could be used as well.)
- This is correlated with how simple the model itself is.
- Completeness: how accurately it describes the model's behaviour.
- Level / Mereological: High level or lower level; coarse grained or detailed; selection of parts and functions.
- Internal or external
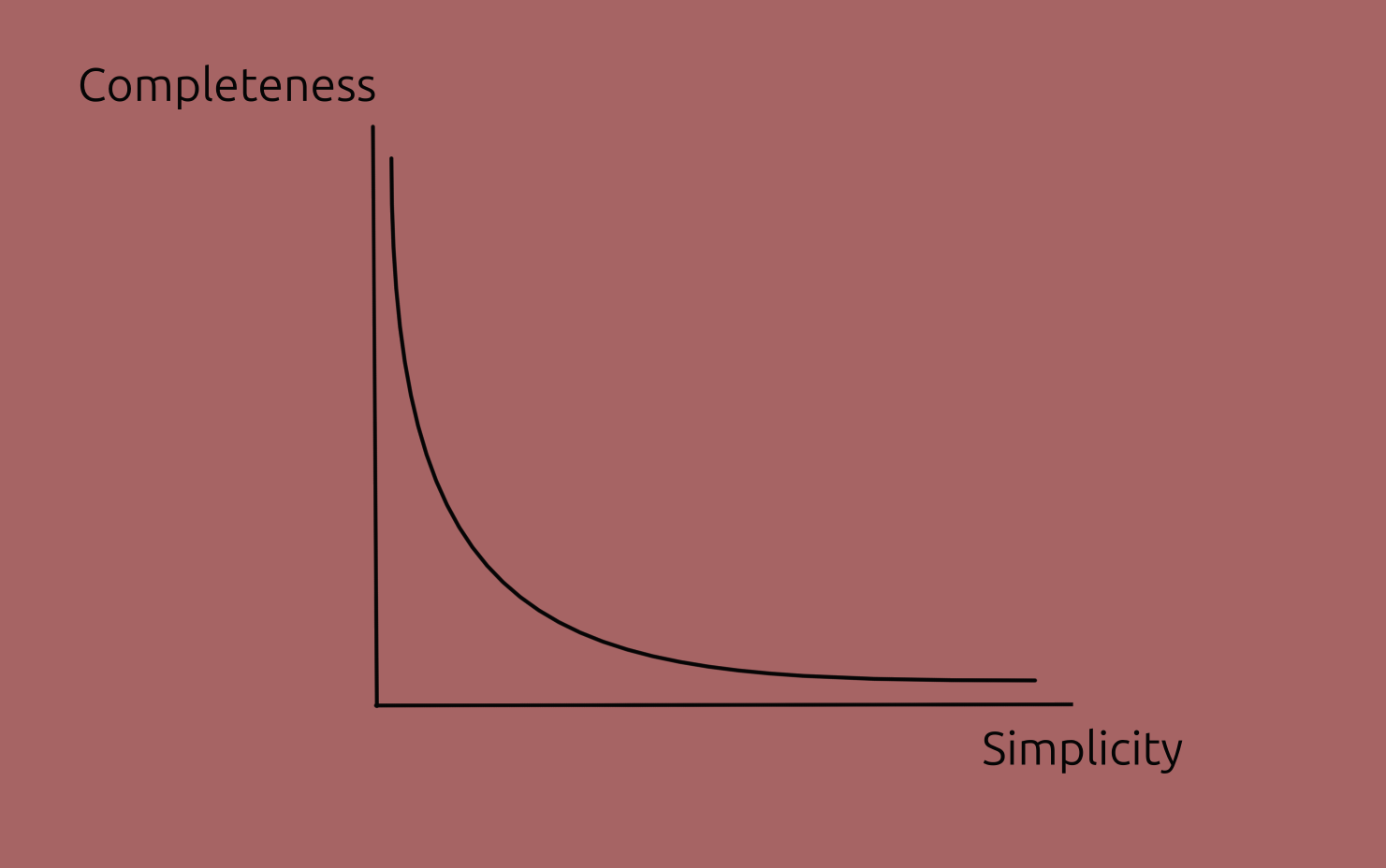
Completeness v. Simplicity tradeoff.
This trade-off isn't universal but just a common case, particularly in deep learning; some other models are straightforward, in which case both characteristics can be high.
Predictive power
Predictive power is a characteristic of a model, not of an explanation of a model. Yet, it is correlated to the characteristics given earlier: more predictive models tend to be more complex making harder to explain them.
The reason to include it here is that predictive power plays an important role deciding which model to use.
In the image below, note that understandability replaces simplicity, and correctness replaces predictive power.
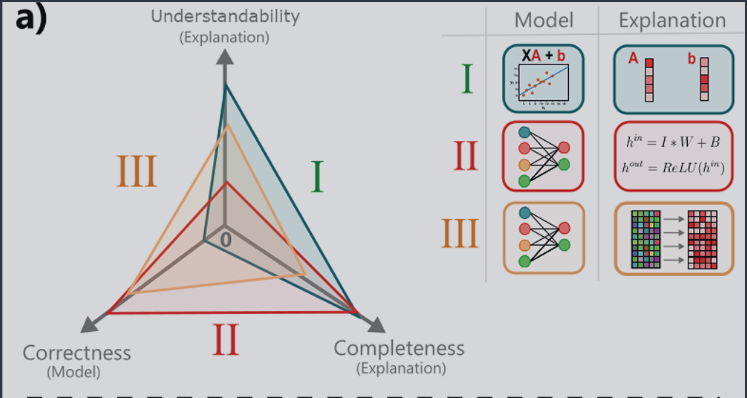
Image from paper under CC-BY-SA 4.0
Let's now look at some actual methods.
Sources:
- A Unified Approach to Interpreting Model Predictions (2017),
- Explaining Explanations: An Overview of Interpretability of Machine Learning (2018),
- [Producing radiologist-quality reports for interpretable artificial intelligence][radiology] (2018),
- The perils and pitfalls of explainable AI: Strategies for explaining algorithmic decision-making (2021) this paper is a particularly short and amenable introduction;
- Interpretable and Explainable Machine Learning for Materials Science and Chemistry (2022),
- Blog Posts: What is Explainable AI? (2022) and from IBM,
- A Perspective on Explainable Artificial Intelligence Methods: SHAP and LIME (2024).
Linear Combination of Features
An important set of methods are linear approximations () to the original (). Mathematically:
s are the effect of each binary feature in the output. Clarifications:
- s do not belong to , but to the approximation ,
- Two complex models trained with same data likely have different s,
- Methods don't protect from a biased model.
Minor note: Linear Combination (of features) is easier for me to remember; but the unified framework paper calls these Additive Feature Attribution Methods.
Best coefficients?
Existing linear combination methods (e.g. SHAP, LIME) calculate s differently, in turn yielding different coefficients. But...which one obtains the best coefficients ? A definition of best is needed.
The Unified Framework proposes that models should have local accuracy, missingness, consistency. With these requirements, they show that Shapley values are the best coefficients. Other methods violate some of these 3 properties.
Technicalities aside, the authors argue these coefficients are more intuitive for humans.
Method: SHAP
SHAP stands for SHapley Additive exPlanations.
The exact Shapley values result from an expensive combinatorial (A value for n-person games, 1952). Approximations to the exact formula can be made, with extra assumptions, which may not hold!!:
- Assumption 1: Feature independence (implies non-multicollinearity).
- Shapley sampling values method,
- Quantitative Input Influence,
- Plus assumption 2, model linearity: Kernel SHAP (LIME + Shapley values)
- Assumption 2, model linearity: Shapley regression values.
SHAP provides both global (average across inputs) and local (for a given input).
Method: LIME
Local Interpretable Model Agnostic Explanation (LIME) and Generalised Linear Models (GLMs).1
For LIME, the coefficients are found minimising an objective function. The coefficients resulting from the optimisation do not necessarily obey the 3 desired properties listed earlier.
Assuming feature independence and model linearity, the objective function can be modified and the SHAP values obtained through weighted linear regression (no slow combinatorics). This is called Kernel SHAP, and obeys the 3 properties listed earlier.
Fixes
- Normalised Moving Rate (NMR): tests the stability of the list against the collinearity. Smaller NMR means more stable ordering.
- Modified Index Position, in the paper's words:
[MIP] works similarly to NMR by iteratively removing the top feature and retraining and testing the model. Thereafter, it examines how the features are reordered in the model which implies the effect of collinearity.
These two methods (MIP, NMR) can be useful both in having a reliable sorting of features, and on selecting one —most stable— of several methods.
Definition of a few concepts
Aside: Collinearity and Non-linearity
Multicollinearity: one feature is a linear combination of one or more other features. For example, ; assuming linear independence would be an error. In the paper's words:
Indeed, some features might be assigned a low score despite being significantly associated with the outcome. This is because they do not improve the model performance due to their collinearity with other features whose impact has already been accounted for.
Non-linearity: output changes are not proportional to input changes. For example is non-linear, and fitting a line to it would be inaccurate. Some SHAP models can model this correctly.
Let's now look at other methods.
-
Local in the name refers to being for a particular input, not Global which would be general. ↩
Explainable AI - Methods II
Other Explainability Methods
-
Saliency Maps: visually show which features are most important in a particular prediction. They can be generated for 1D, 2D and ND inputs. For example, here is for radiology:
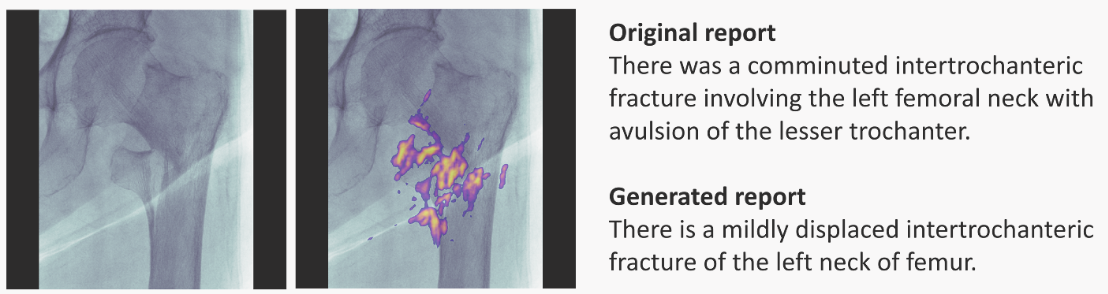
Left-most: input image; next: input + saliency map; right-most: doctor's annotation (top) and RNN-model generated annotation (bottom). Image taken from paper.
-
Validity Interval Analysis: another technique fitting the NN behaviour to try to extract explanations.
-
Dimensionality Reduction: Principal Component Analysis, t-SNE, Dimensionality Reduction, Independent Component Analysis, Non-negative Matrix Factorisation can all help as well.
Explanation-producing Architectures
Architectures designed to make explaining part of their operation easier.
-
Using Explicit Attention: An attention layer/mask learns how parts of an input embedding pay attention to other parts. The layer is somewhat interpretable. In chemistry, it could learn which atoms connect (or pay attention to) other atoms.
-
Dissentangled Representations:
Disentangled representations have individual dimensions that describe meaningful and independent factors of variation.
—Explaining Explainability (2018). Examples of architectures are -VAE, INFOGan, capsule networks.
Discovering Inorganic Solids
These are some of my opinions and ideas after reading two papers by Rosseinsky group:
- Discovery of Crystalline Inorganic Solids in the Digital Age (2025).
- Element selection for crystalline inorganic solid discovery guided by unsupervised machine learning of experimentally explored chemistry (2021)
Introduction
In solid-state chemistry, some elemental compositions (phase fields) are more likely to lead to isolable compounds than others.
Deep learning models can help differentiate between these two groups, and lead researchers to the promising areas. The models can be trained for this task with data from ICSD, the Inorganic Crystal Structure Database.
Such models would improve the allocation of resources when exploring new phase fields.
Searching for new compounds
Some definitions will be used:
- Phase field: the elements selected. Can be thought as the labels for cartesian axes.
- Composition: the values or ranges of values in each axes. Once we have the axes' labels we can explore values computationally.
We can search for compounds by analogy and by exploration, characterised in the table below:
| Method | Starting Point | Concept | Success Rate |
|---|---|---|---|
| By analogy | Parent Compound | Change composition, same structure | Higher |
| By exploration | Structural Hypothesis / Idea | Try composition and structure | Lower |
Analogy Based Search
The analogy-based search involves:
- Starts from a naturally occuring mineral, or previously discovered structures,
- Change its composition retaining the crystalline structure. For example, can be expanded by analogy to , conserving the crystalline structure.
With respect to analogy-based search, the paper notes:
(...) it is straightforward to expand known structures by analogy through substitution, but the initial identification of such structures, which cannot be by analogy, is an entirely different question (...)
And usefully,
The properties of the analogy-based materials can be superior to those of the initial discovery (...)
Exploratory Search
The ML-aided exploratory-search involves:
- Human selects elements or phase field e.g. , ,...
- A VAE decodes the seed-input into similar compounds (nearby in latent space).
- The reconstruction loss is used as a ranking metric for the generated compounds.
- Computationally search in composition-space (Crystal Structure Prediction, CSP), find low-energy probe structures, e.g. .
- Can use physical constraints (like max n of atoms).
- Calculate thermodynamically stable1 probe structure (this step is complex). Hints experimentalists of promising region.
- Try synthesis, and find somewhat similar structures to the computationally suggested one.
We can describe the exploration steps as a flow as well:
---
config:
flowchart:
htmlLabels: true
---
flowchart LR
A(("`**Input Phase**
(e.g Na-Ca-O)`")) --> B(VAE)
B -- "`**Ranked Phases**`" --> D(Distance Metric)
D -- "`**Compositions**`" --> F(Thermodynamics)
F -- "`**Probe**`" --> H(Try synthesis)
style A fill:#123456,color:#f4f4f4,stroke-width:0px
-
With respect to the convex hull. ↩
Discovering Inorganic Solids
In depth workflow
Dataset
The atom descriptors are taken from a atom-property database and include atomic weight, valence, ionic radius, and others.
The 4-element crystals are selected from ICSD, and only the elements are retained. For example, would become for training.
The data is scaled 24 fold by performing all possible permutations of 4 elements, i.e. 4! (factorial). This enhances learning, reduces overfitting.
Architecture: Variational Autoencoder (VAE)
Here the emphasis is on exploiting a pattern and not on interpretability, the human expert evaluates the compounds afterwards.
An autoencoder consists of two parts, an encoder, and a decoder. The overall task is to reconstruct the original vector from the compressed representation.
The encoder compresses the 148 vector into a 4D vector (latent vector), and the decoder decompresses it into 148D. The euclidean distance is then computed as a measure of error, and the gradient is used to correct the weights.
Since the model is trained only on phase fields that lead to isolable materials, it is biased towards those compounds.
Just like a single-class classifier using cat-only images, the VAE only sees positive instances, and no learning comes from predicting negatives.
Inference Stage
Input structures are passed with a bit of noise each time and the reconstruction loss is used to rank them for synthetic exploration.
A larger reconstruction loss means the phase is less likely to be synthesizable, since it learn to reconstruct only synthesizable regions.
The rank will also tell how different the compound is to the original.
Example of Results
After VAE ranking, the decision to explore Li-Sn-S-Cl phase field was based on the high conductivity of a related ternary field Li-Sn-S.
The following image shows calculations performed, each a tripod, in a red background. Dark red represents little enery barrier from the convex hull, bright red the opposite.
Most solids found by the group or by others are in dark areas of the plot with tripods overlaying.
The magenta point A in the image is the new phase found, not far from the probe structure which was .
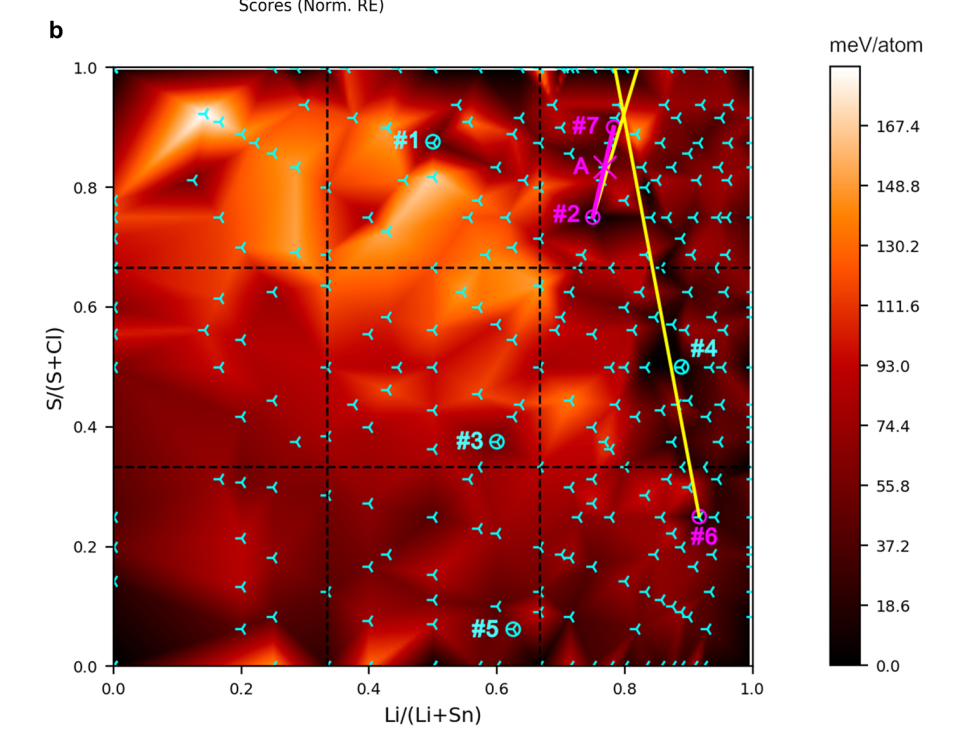
Image from Original Paper under CC-BY-SA 4.0
Machine learning for molecular and materials science
Useful snippets from the Perspective paper Machine learning for molecular and materials science.
Representations of atoms, molecules, materials
The process of converting raw data into a format more suitable for an algorithm is called feature engineering.
The more suitable the representation of the input data, the more accurately can an algorithm map it to the output data.
Selecting how best to represent the data may require insight into both the underlying scientific problem and the operation of the learning algorithm, since it is not always obvious which choice of representation will give the best performance
Some representations:
- Coulomb Matrix: atomic nuclear repulsion information.
- Graphs: connectivity of molecules.
- String representations: SMILES, SELFIES,..
- Solid-state unit-cells: Representations based on radial distribution functions, Voronoi tessellations, and property-labelled materials fragments (...)
In the solid-state, the conventional description of crystal structures by translation vectors and fractional coordinates of the atoms is not appropriate for ML, since a lattice can be represented in an infinite number of ways by choosing a different coordinate system.
Areas
They see possible impact in a few areas:
- Synthesis: retrosynthesis, crystallisation predictions, etc.
- Characterisation: for example analysing images (CV)
- Modelling: reducing time and improving accuracy of calculations
- Drug discovery, Drug design, Inverse Design, Property Prediction,..
Algorithms
- Naive Bayes:
Bayes’ theorem provides a formal way to calculate the probability that a hypothesis is correct, given a set of existing data.
- Nearest Neighbour:
In nearest neighbour (k-NN) methods the distances between samples and training data in a descriptor hyperspace are calculated. k-NN methods are so-called because the output value for a prediction relies on the values of the k nearest neighbours, where k is an integer.
- Decision Trees:
are flowchart-like diagrams used to determine a course of action or outcomes. (...) with branches indicating that each option is mutually exclusive. (...) Both root and leaf nodes contain questions or criteria to be answered.
-
Kernel methods are a class of algorithms; whose best known members are the support vector machine (SVM) and kernel ridge regression (KRR).
-
Artificial neural networks (ANNs) and deep neural networks (DNNs). (...) Learning is the process of adjusting the weights so that the training data are reproduced as accurately as possible. (...) The values of internal variables (hyperparameters) are estimated beforehand using systematic and random searches, or heuristics.
Best Practices
TL;DR from the paper "Best practices in machine learning for chemistry" (2021), a very similar paper by the same authors is "Machine learning for molecular and materials science".
- For Datasets
- Ensure dataset remains available, and is version-tagged (they change)
- For home-made or mixes, explain the process of generation
- Describe any data curation, balancing, augmentation, and so on.
In the Best Practices paper they say:
For reasons of reproducibility, it is crucial that these databases use some mechanism for version control (e.g. release numbers, Git versioning, or timestamps) as part of the metadata and maintain long-term availability to previous versions of the database.
-
For Representations
- Try more than one, compare
- Use very basic ones as baseline representation to compare (example random or one-hot)
-
Justify Model
- More complex isn't always better
- Compare to baselines (mean for regression, most common class for classification)
- Compare to very simple models and to SOTA
- Any interpretability we can offer?
-
Evaluate Model
- Have 3 separate datasets: for training and optimising, for evaluating during training and detect overfitting, and testing for testing which should represent where it will be applied (should test what we want it to succeed on).
- Test extrapolative learning: leave out some class entirely, or train until a cutoff date and evaluate with dates after that.
- Test intrapolative learning: with varied test sets
- Mindful of shorcut learning (have varied test dataset).
-
Reproducibility: Results and code must be made available and reproducible
They also state:
In all reports, remember to cite the methods and packages employed to ensure that the development community receive the recognition they deserve.
They provide a great checklist but since the license is a mess I am not including it here.
Example
Take ElemNet as an example and go through the checklist.
- Database:
- They provide a link, but no timestamped or git version,
- No info on curation or preprocessing (we may assume none was performed).
- Representations:
- They justify and compare the results to baselines.
- Model:
- Describe why is new and useful idea,
- Describe architecture.
- Evaluation:
- Show training and different hyperparameters,
- Studied which compounds model is accurate vs not.
- Reproducibility: Results and code are available.
The comparison would be improved if they also ran a deep learning model with human-made descriptors.
They also included other useful statistics like inference time.
Databases and Benchmarks
Bear in mind when using databases what this Machine learning for molecular and materials science states:
Data may require initial pre-processing, during which missing or spurious elements are identified and handled.
Identifying and removing such errors is essential if ML algorithms are not to be misled by their presence.
- The Machine Learning for molecular and materials science aggregates many DBs and such in tables at the end
- Pillong, M. et al. A publicly available crystallisation data set and its application in machine learning. CrystEngComm (2017).
- ICSD: Inorganic Crystal Structure Dataset
- Jain, A. et al. The materials project: a materials genome approach to accelerating materials innovation.
- The materials project database https://materialsproject.org/
- Benchmarking materials property prediction methods: the matbench test set and automatminer reference algorithm.
- Matbench benchmark: https://hackingmaterials.lbl.gov/automatminer/datasets.html
- Materials design and discovery with high-throughput density functional theory: the open quantum materials database (OQMD)
Also ElemNet lists materials-and-properties' databases (experimentally observed and hypothetical):
DFT calculations have offered opportunities for large-scale data collection such as the Open Quantum Materials Database (OQMD), the Automatic Flow of Materials Discovery Library (AFLOWLIB), the Materials Project, and the Novel Materials Discovery (NoMaD); they contain DFT computed properties of of experimentally-observed and hypothetical materials. In the past few decades, such materials datasets have led to the new data-driven paradigm of materials informatics
ElemNet describes OQDM as well (bold is mine):
OQMD is an extensive high-throughput DFT database, consisting of DFT computed crystallographic parameters and formation enthalpies of experimentally observed compounds taken from the Inorganic Crystal Structure Database (ICSD) and hypothetical structures created by decorating prototype structures from the ICSD with different compositions.
CompChem Map
This is a draft of areas I'd like to organise in some taxonomy.
Finding Useful Molecules
- Get a Materials Database(s), either method
Method 1: Direct (Compounds to Properties)
- Use DFT to guide towards one that fits the requirements (slow if we have billions of compounds.),
- Or use the DB to train a NN to make predictions (needs labelled data for training)
- Or similarity metrics to find new (similar) molecules.
- ...
Method 2: Inverse (Properties to Compounds)
- Use the gradient to update embedding.
- Maximise or Minimise the needed properties.
The paper's approach is more towards a Direct method. It is a method to generate embeddings that can then be used to train a neural network to predict properties.
This can be arranged (not very tidily) in a chart:
---
config:
flowchart:
htmlLabels:false
---
flowchart TB
A[("Compounds Database")]
subgraph Direct["`**Direct**`"]
direction LR
B("`Electronic Structure
Predictions`")
C("`Train NN on DB
(with labelled data)`")
D("`Use similarity metrics to
find nearby candidates`")
end
subgraph Inverse["`**Inverse**`"]
direction TB
E("`Train VAE to create
smooth surface`")
E --> F("`Link MLP to
latent vector`")
F --> G("`Minimise or Maximise pps.
by changing vector`")
end
A --> Inverse
A --> Direct
Unsupervised-Learning of Representations of Atoms
Other investigations of unsupervised learning of machine representation of atoms are:
- Zhou, Q. et al. Learning atoms for materials discovery. (2018).
- Tshitoyan, V. et al. Unsupervised word embeddings capture latent knowledge from materials science literature. (2019).
- Chakravarti, S. K. Distributed representation of chemical fragments. (2018).
- Butler K. et al. Distributed Representations of Atoms and Materials for Machine Learning. (2022).
Supervised-Learning of Representations of Atoms
- Jha, D. et al. ElemNet: deep learning the chemistry of materials from only elemental composition. (2018).
- Goodall, R. E. & Lee, A. A. Predicting materials properties without crystal structure: deep representation learning from stoichiometry. (2020).
Visualising High Dimensional Data
- PCA
- Dimensionality Reduction
- t-SNE
Reference of some of the techniques, book cited in some papers: 18. Hastie T, Tibshirani R, Friedman J (2001) The Elements of Statistical Learning, Springer Series in Statistics (Springer, New York).
Queries
- How to build useful machine-representations of atoms?
- would just be a 1D vector embedding, likely of little use. But are there taxonomies of representations (including a matrix per atom?)
Missing
- Splits for datasets, validation / testing.
- Balancing datasets.
- Evaluation metrics, loss functions,
- ROC classification performance metric
Ideas
-
Train and show the results of atoms for vectors in a website? With some button to load each dataset?
-
List of examples of successful applications of ML in chemistry
- Anything for which there are useful datasets from experiments or from calculation:
- orbital energies,
- geometries,
- formation energy,
- energy band-gap
- Anything for which there are useful datasets from experiments or from calculation: